江南大学尹健教授团队在Chemical Science上发表的题为《Tandem activated caged galactoside prodrugs: advancing beyond single galactosidase dependence》的文章,该研究提出并系统验证了一种全新的双刺激响应型半乳糖苷类前药(TACGs)策略,以克服传统β-半乳糖苷酶(β-gal)依赖前药在特异性释放方面的限制。

全文速览:
β-半乳糖苷类前药因可被某些癌细胞中高表达的β-半乳糖苷酶(β-gal)激活,过去三十年来一直被探索用于抗癌治疗。然而,β-gal在体内的分布缺乏足够的特异性,难以确保药物能够精准地在肿瘤部位释放。为解决这一问题,本文利用β-gal对底物高度严格的特异性,选择天然存在于半乳糖上的羟基作为前药修饰位点,开发出一类新型的串联激活“笼式”半乳糖苷(Tandem activated caged galactoside,TACG)前药。这类前药除了需要β-gal,还需额外刺激才能激活,实现更精准可控的按需释药。研究证明,将不同的“保护基”连接在半乳糖的6-位羟基上,可以让这些半乳糖苷对β-gal酶水解产生抗性。用一种光敏保护基——4,5-二甲氧基-2-硝基苄基(DMNB)修饰,即6-O-DMNB半乳糖苷前药,包括康布瑞他汀A4(combretastatin A4)和8-羟基喹啉(8-hydroxyquinoline)等药物分子,结果表明它们在紫外光和β-gal共同作用下的抗癌活性。本研究为构建“双刺激响应”的半乳糖苷前药提供了一种简便、高效、且具有广泛适用性的策略,有望推广至多种糖苷类前药的设计与开发,推动基于碳水化合物的药物研发进展。
全文详解:
研究背景:
笼形前药(Caged prodrugs)通过对药物分子中的关键官能团(如羟基或氨基)进行化学修饰,使其失活或活性大幅降低,待特定刺激(如光、氧化还原、低pH、酶切或生物正交反应)触发后释放活性药物。这种策略广泛应用于药物化学与化学生物学中。由于癌细胞代谢异常,常表现出特定酶活性的升高,因此可设计酶响应型前药,使药物仅在癌细胞中被激活,提高化疗的选择性。β-半乳糖苷酶(β-gal)在某些癌细胞(如卵巢癌)中表达上调,因此β-半乳糖苷前药在抗癌中有较大应用前景。自1994年以来,研究者开发了多种β-gal响应前药,包括氟尿嘧啶、duocarmycin、多柔比星、MMAE、康布他汀A4、吉西他滨等。其中一些还结合靶向配体或成像探针,实现治疗-诊断一体化。然而,β-gal并非癌细胞特有,在多种正常细胞(如巨噬细胞、神经元、肝脏、肠黏膜等)也有表达,甚至在衰老细胞中也呈高表达,增加了非特异性毒性的风险。为提高激活精准性,研究者提出双刺激响应前药的设计策略,即需两个不同刺激同时存在才能激活药物,相当于“AND门”逻辑。目前已有如组蛋白去乙酰酶/半胱天冬酶L、磷酸酶/转环辛烯、活性氧/酪氨酸酶等组合的双刺激前药,但应用于糖苷类前药的还很少。理论上,构建双刺激响应β-gal前药可采用两种方法:一是将糖苷和另一遮蔽基团分别连接于药物不同部位,但这需药物有两个锚点,设计限制大;二是将额外遮蔽基团插入在糖苷和药物之间,设计更复杂且难以推广。本研究提出一种普适性的双刺激响应β-gal前药设计策略:在β-半乳糖苷的羟基上引入遮蔽基团,只有在该基团被移除后,β-gal才能识别并释放药物。这种前药被称为串联激活型笼形半乳糖苷(TACGs),设计理念源于β-gal对底物结构的高度选择性——哪怕羟基轻微变化也会抑制其水解活性。

研究创新点:双刺激响应策略(Dual-Trigger Strategy)
· 提出一种串联激活的“笼式”半乳糖苷前药(Tandem Activated Caged Galactosides, TACGs):
o 第一刺激:如光照(使用DMNB光敏遮蔽基团);
o 第二刺激:β-galactosidase水解β-半乳糖苷键。
· 只有在两个刺激同时存在时,药物才被释放,类似逻辑“AND门”控制,大幅提升了前药释放的精准性和安全性。具体而言,第一个刺激为外源性光照,利用光敏遮蔽基(如4,5-二甲氧基-2-硝基苯基,DMNB)阻断半乳糖分子的关键羟基位点,从而抑制β-gal对糖苷键的识别和水解;第二个刺激为内源性酶β-gal,当遮蔽基在光照下脱落后,原位羟基暴露,β-gal才可识别并断裂糖苷键,最终释放活性药物。这种“光+酶”双激活机制使前药系统实现类似“AND逻辑门”的精准控制,避免了在单一刺激下的非特异性药物释放,有望大幅提升抗癌治疗的选择性与安全性。
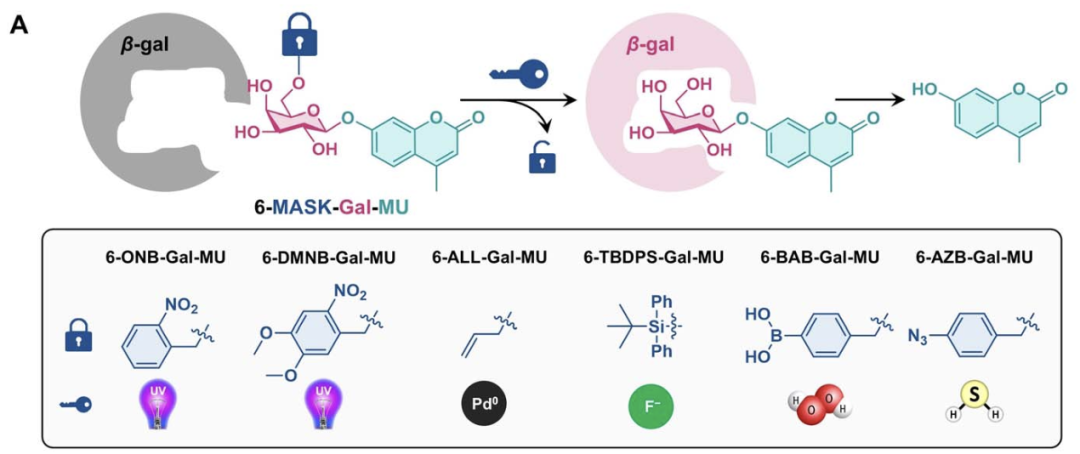
设计与合成策略:利用半乳糖羟基修饰锁定酶识别
· 以β-D-半乳糖的2-, 3-, 4-, 6-位羟基作为修饰位点,通过接入光敏遮蔽基(如DMNB)抑制β-gal识别和水解。
· 在激发光照作用下,遮蔽基脱落,羟基恢复,从而β-gal可识别并水解糖苷键,最终释放活性药物。
· 该策略与传统“先糖后药”策略不同,强调通过糖的羟基结构控制酶活性,更具普适性和可拓展性。
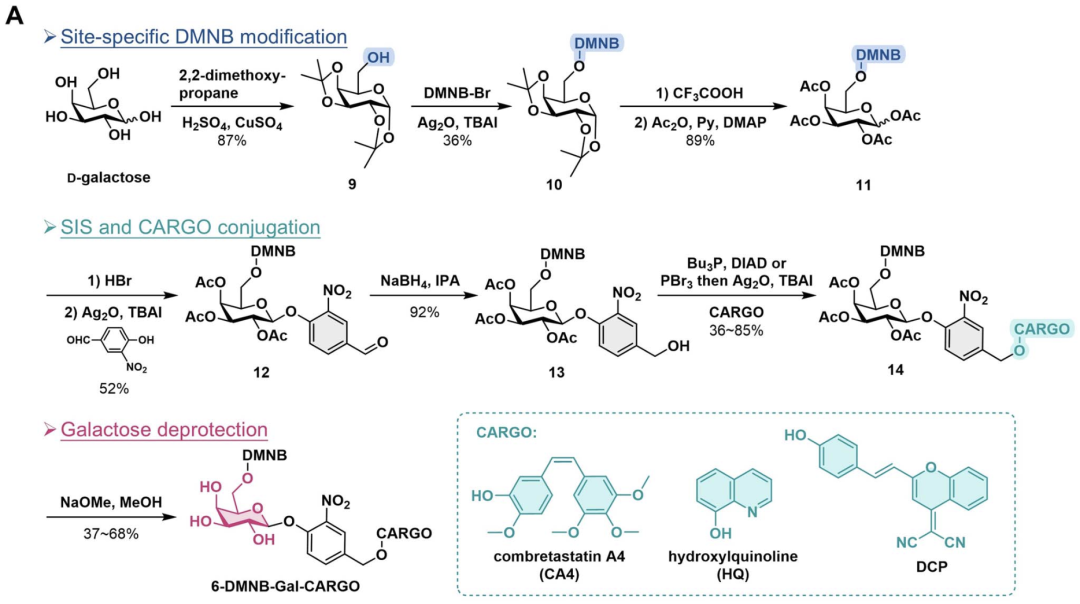
代表性化合物与结构活性关系(SAR):药物实例
· 研究以两个已知药物为例验证策略有效性:
o 康布瑞他汀A4(CA4):抗微管药物;
o 8-羟基喹啉(8-HQ):具有金属螯合和抗癌活性。
· 合成多个位点(O2, O3, O4, O6)DMNB修饰的TACGs,系统比较它们在光照+β-gal条件下的药效。
SAR结果:在所有构建的异构体中,O2位DMNB修饰的TACG结构显示出最佳的性能:不仅在光照条件下遮蔽基能够高效脱除,而且释放后的产物能被β-gal快速识别和水解,展现出最优的双刺激响应特性。以CA4为例,其O2-DMNB修饰的TACG前药在紫外光照射+β-gal共同作用下迅速释放出母体药物,显著抑制细胞微管聚合,具备强烈的UV依赖性抗癌活性。
总结&展望:
本研究建立了一种简洁、模块化、具有高度适配性的双刺激响应前药构建平台,为糖苷类前药的精准释放提供了新范式。该策略不仅适用于现有的β-gal靶向前药,也可通过更换遮蔽基(如响应ROS、pH、还原环境、特定酶等)实现对不同病理环境的适配;同时,通过选择不同羟基位点修饰,可以进一步调控药物的理化性质、膜通透性、血浆稳定性等关键药代动力学参数。更重要的是,这一策略并不局限于β-gal系统,理论上可拓展至其他糖苷酶介导的药物激活平台,具有广泛的应用前景,特别适用于癌症、炎症、自身免疫疾病等病理状态中存在微环境变化的精准药物递送领域。
参考文献&原文链接:
https://pubs.rsc.org/en/content/articlepdf/2025/sc/d5sc00722d


 微信服务号
微信服务号
 微信订阅号
微信订阅号
 微信订阅号
微信订阅号